{kind=link}
Susan was nonetheless a baby when she first suspected one thing may be incorrect together with her mom. A cup or plate would usually crash to the ground by chance when her mom was serving dinner or washing up dishes. “She was, she would have said, ‘clumsy’, but she wasn’t really clumsy,” says Susan. “Her hands had beautiful, glamorous movements, which I now recognize as early HD.”
Huntington’s illness (HD) is an inherited situation that causes widespread deterioration within the brain and disrupts pondering, behaviour, emotion and motion. The illness often begins in midlife, with refined modifications corresponding to temper swings and problem in staying targeted. As it progresses, folks develop dementia and an incapability to talk or transfer.
Susan, who requested that her final identify be withheld to guard her privateness, vividly remembers the day she learnt that her mom had the illness. It was the spring of 1982, and her mom had been admitted to a hospital due to her excessive exhaustion, frequent falls and irregular actions. There was no genetic check for the situation on the time, so she underwent a sequence of assessments. Her neurologist gathered your entire household right into a room to interrupt the information. “He told us that our mother had Huntington’s disease,” recollects Susan. “And that there’s no treatment and it can be wiped out in a generation if you just don’t breed.”
Those blunt phrases had a profound influence on the lives of Susan and her siblings: her brother determined by no means to get married, and her sister selected to be sterilized. For Susan, nonetheless, these choices have been out of attain: she was pregnant when she acquired the information.
Susan says that she and her husband “couldn’t decide what was the right thing to do”. One thought, particularly, was that “if we have the child, then that child will have this same decision when they grow up”, she says. “And it seemed so cruel.” Ultimately, the couple made the heart-wrenching option to terminate the being pregnant.
The gene concerned in Huntington’s, known as HTT, codes for a protein known as huntingtin. The defective model of the gene repeats a brief piece of its sequence — the nucleotide mixture CAG — too many instances. Unlike some genetic circumstances, during which an individual will develop a illness provided that they’ve two defective copies of a gene, only one copy of the HTT mutation is sufficient to result in Huntington’s, and carriers of the mutation have a 50% probability of passing it on to their kids. Years after Susan’s mom handed away, the three siblings found that they’d all inherited the illness.
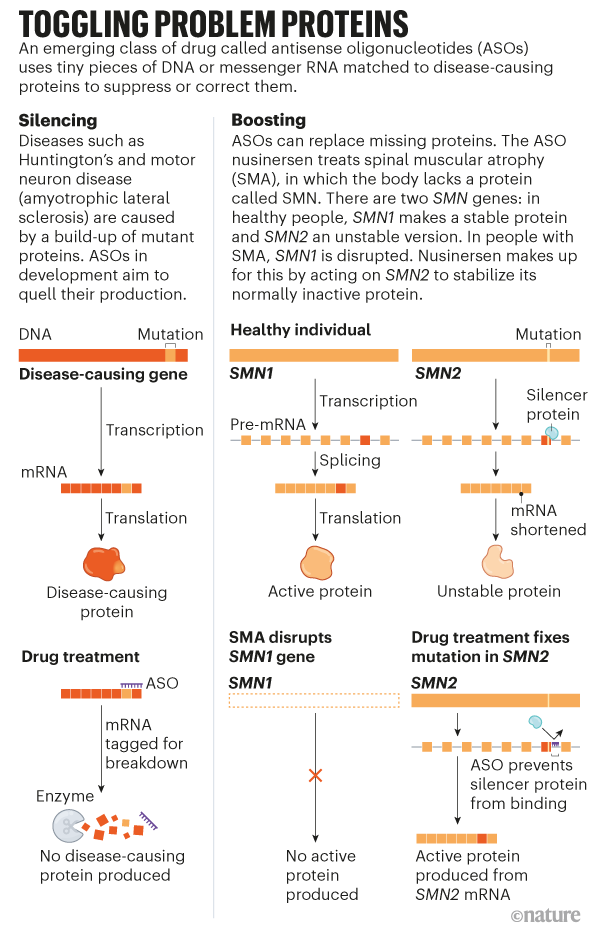
There are not any remedies out there to cease or sluggish the development of Huntington’s, though its genetic trigger has been clear since 1993. Most different neurodegenerative diseases additionally lack efficient therapies and, though their genetic roots are much less clear-cut than for Huntington’s, lots of the genes related to circumstances corresponding to motor neuron illness (amyotrophic lateral sclerosis, or ALS), Alzheimer’s and Parkinson’s have been identified for many years. Now, the tide may be turning for treating these sorts of diseases. Many researchers are hopeful about medicine often known as antisense oligonucleotides (ASOs). These are quick strings of DNA or RNA letters which can be designed to cling to specific sequences of RNA made by defective genes, and to rebalance the degrees of proteins they produce — boosting lacking proteins or quashing defective ones (see ‘Toggling problem proteins’).
Credit: Nik Spencer/Nature
The US Food and Drug Administration (FDA) authorized the primary ASO for a neurological illness in 2016, and there has since been an explosion of exercise on this space. The subject has gone from only a handful of medical trials run over the previous 20 years to round a dozen presently below means for varied neurodegenerative diseases — and some have reached their ultimate phases.
Other ASO researchers are transferring past diseases outlined by a single mutation to take a look at circumstances with more-complex genetic underpinnings. This latest progress has made many within the space optimistic about the way forward for the expertise. Don Cleveland, a neuroscientist on the University of California, San Diego (UCSD), and one of many first scientists to research the usage of ASOs for neurological diseases, sees this as only the start. “There’s much, much more coming,” he says.
But progress within the subject has not been utterly easy. At the tip of final month, a large phase III trial was abruptly halted as a result of the good thing about the drug to sufferers didn’t outweigh the dangers. And some researchers have lengthy urged warning round ASOs, owing to the truth that their efficacy in lots of circumstances is unknown and that the way in which they’re delivered — usually by spinal injection — is invasive.
Although the end result of this trial was disappointing, “I don’t think this is a reason for despair,” says Chris Boshoff, a scientific-project supervisor overseeing genetic therapies on the US National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. “There’s still reason to be positive and enthusiastic about what this modality can accomplish.”
Breakthrough for a uncommon illness
Elliot and Janell Lewis’s first youngster, Blakely, was born in 2011 with a uncommon, inherited neurodegenerative illness often known as spinal muscle atrophy (SMA). People with SMA have a mutated type of SMN1, a gene answerable for producing a protein known as survival motor neuron (SMN). The ensuing lack of SMN prevents the brain from with the ability to talk successfully with the physique, resulting in muscle weak point and losing that worsens over time. There are 4 kinds of SMA; the most typical type, SMA1, can also be probably the most extreme. People with SMA1 sometimes present signs shortly after start, and many don’t survive previous the age of two.
Blakely was recognized at three months previous. “That pretty much shattered us,” Elliot says. At the time, there was no therapy, and Blakely handed away at 21 months.
In the spring of 2017, the couple had one other daughter, Evie. Evie additionally had SMA, however she was extra lucky — just a few months earlier than she was born, the FDA authorized an ASO, dubbed nusinersen, the primary ever disease-modifying therapy for SMA. Evie acquired her first dose when she was 12 days previous.
Scientists first acknowledged1 the power of ASOs to focus on RNA in 1978, nevertheless it took a number of many years to reveal their medical potential. Early on, issues corresponding to toxicity and lack of efficiency stymied progress, and plenty of drug firms misplaced curiosity. But researchers at one agency, Ionis Pharmaceuticals (initially named Isis Pharmaceuticals), based mostly in Carlsbad, California, launched key modifications to the medicine’ chemical spine that elevated efficiency in addition to stability, enabling the ASOs to achieve their targets with out being degraded.
The work that led to nusinersen started round 2000 at Cold Spring Harbor Laboratory in New York, the place biochemist and molecular geneticist Adrian Krainer was investigating the mechanisms that led SMN2, one other gene that encodes SMN, to sometimes produce much less viable protein than its counterpart, SMN1. They reasoned that if they might get SMN2 to provide extra protein, it might make up for SMN1 in folks with a mutation in that gene. They knew from others’ work that in nearly everyone, the reason for the issue with SMN2 was an error throughout splicing — the method by way of which strands of RNA are snipped and processed into directions for making proteins. That causes a bit of SMN2’s code to be skipped.
Krainer’s crew zoomed in on the proteins that bind to the RNA strand and trigger the phase to be missed, hoping to cease them interfering within the means of producing full SMN proteins. In 2004, Krainer started collaborating with Frank Bennett, a pharmacologist and one of many founding members of Ionis Pharmaceuticals. Together, they pinpointed an ASO that would bind to the strand and conceal the phase from the proteins that may silence it, enabling the manufacturing of practical SMN2.

Four-year-old Evie Lewis performs within the household residence in Ogden, Utah.Credit: Kim Raff for Nature
That compound, nusinersen, entered medical trials in 2011. The outcomes have been so promising that the part III trial in infants with SMA was terminated early: sufferers who acquired the drug have been more likely to fulfill their motor milestones and survive than have been those that acquired a placebo3.
So far, greater than 10,000 folks worldwide have acquired nusinersen (Spinraza), which Ionis licensed to drug maker Biogen, based mostly in Cambridge, Massachusetts, in 2016. The drug has drastically altered the course of the illness: infants with SMA who obtain it shortly after start are now not dying throughout the first years of life. Nowadays, “conversations [with families] don’t just end with, ‘We’re going to do everything we can, but your baby’s going to die’,” says Russell Butterfield, a paediatric neurologist on the University of Utah in Salt Lake City (Butterfield has acquired consulting funds from Biogen). “Instead, that conversation switches to, ‘We have this new drug, it’s absolutely amazing. We need to get it in as soon as possible’.”
Evie Lewis, now 4 years previous, receives a dose of Spinraza by a lumbar puncture each few months, and she or he just lately had her 15th injection. Although she nonetheless faces some points, corresponding to having to eat by way of a feeding tube, she is ready to stroll, run and climb — issues that Blakely was by no means capable of do, Elliot says.
A packed pipeline
Following the success of nusinersen, researchers started to deal with different diseases related to clearly outlined genetic mutations, corresponding to Huntington’s. That led to the drug tominersen, which was developed by Ionis and licensed for medical testing to the pharma firm Roche in Basel, Switzerland. It is believed to work by focusing on CAG repeats on the RNA strand produced by each the conventional and defective HTT genes, and tagging them for destruction by an enzyme known as RNase H1. The outcomes of a part I/II medical trial, which have been printed in 2019, revealed that tominersen lowered concentrations of the mutant model of huntingtin within the cerebrospinal fluid, with out inflicting any severe unintended effects4.
The success of the early Huntington’s trial caught the eye of neurodegeneration researchers, as a result of tangles of protein are a key function of many such issues. “There was a lot of excitement about this, because it really opened up the doors to be able to do antisense trials for other neurodegenerative diseases where build-up of a toxic mutant protein plays a role,” says Sarah Tabrizi, a neurologist at University College London, who led the part I/II trial of tominersen.
But an surprising announcement on the finish of March dealt an enormous blow to the Huntington’s group. A part III trial of tominersen involving 791 individuals from 18 nations was terminated early on the recommendation of an unbiased committee of consultants, who had performed a deliberate overview of the info. A press release from Roche mentioned that no new security considerations had emerged, however that the drug’s potential advantages didn’t outweigh the dangers. Until extra particulars are printed, it’s not potential to say what went incorrect, says Tabrizi.
Drugs that work in an analogous option to tominersen are nonetheless in play for different issues with related causes. Some circumstances of ALS, as an illustration, are attributable to an excessive amount of of a mutant protein, and a handful of ASOs for these types of the illness are in medical trials. The furthest alongside is tofersen, an ASO developed by Ionis to deal with an inherited type of ALS. Tofersen is now being examined in a Biogen-sponsored phase III trial.
Claudia Testa, a neurologist at Virginia Commonwealth University in Richmond, says that there are distinctive challenges that include decreasing the degrees of a mutant protein, as tominersen and tofersen do, in contrast with boosting a lacking one, as nusinersen does. Several protein-lowering methods really cut back ranges of each good and dangerous variations of a protein. Scientists don’t but know the long-term results on the diseases involved, and it’s not clear if this was the difficulty within the part III trial of tominersen. The drug for SMA is doing one thing essentially completely different, “so it doesn’t predict efficacy for the other diseases — and that’s a painful truth”, says Testa.
To keep away from this downside, some ASOs are aimed squarely at mutant proteins. One biotechnology firm, Wave Life Sciences in Cambridge, Massachusetts, is testing a technique that targets tiny mutations that typically happen alongside the CAG repeats on simply the mutant copy of HTT. The intention is to depart ranges of wholesome huntingtin comparatively intact. But the drug would work solely in a subset of individuals with Huntington’s who carry these mutations. Furthermore, that distinction may be recognized solely with an exhaustive sequencing technique that isn’t routinely carried out within the clinic, says Testa. (Testa has acquired consulting charges from Wave Life Sciences.)
A poisonous model of the HTT protein, which causes Huntington’s, forming clumps (shiny inexperienced).Credit: Ok. W. Drombosky et al./Neurobiol. Dis.
More just lately, researchers have began testing ASO-based therapies for more-common neurodegenerative circumstances, corresponding to Parkinson’s and Alzheimer’s. The overwhelming majority of circumstances aren’t linked to a selected genetic mutation, and these issues are rather more prevalent than are inherited diseases. The ASO for Alzheimer’s aims to lower levels of tau, a protein that builds up into poisonous tangles within the brain. For Parkinson’s, the objective is to lower the α-synuclein protein, which aggregates into pathological clumps often known as Lewy our bodies.
But for neurogenerative diseases corresponding to these, a number of genes in a community are more likely to be concerned, says Kevin Talbot, a neurologist on the University of Oxford, UK, who can be concerned in a forthcoming trial of an ASO for ALS. It’s unclear how a change to at least one gene within the community would have an effect on the remaining, he says. (Talbot has beforehand served on scientific advisory boards for Roche and Biogen.)
Another problem, based on Talbot, is that these medicine presently should be delivered utilizing repeated lumbar punctures to achieve their targets within the central nervous system. Before ASOs may be utilized to a wider vary of diseases, it is going to be essential to discover a option to get these medicine previous the blood–brain barrier in order to ship them much less invasively, Talbot says. “There’s a whole list of things that have to be done before we get too triumphalist.”
Change of identification
Studies in mice counsel that the ASOs of the longer term might have much more highly effective makes use of within the brain: changing misplaced neurons.
Last yr, Xiang-Dong Fu, a cell biologist at UCSD, and his colleagues demonstrated that it’s potential to make use of ASOs to transform non-neuronal brain cells known as astrocytes into neurons5. The crew injected an ASO right into a area of the mouse brain from which neurons are misplaced in Parkinson’s. Once there, the drug activated a community of genes that prompts astrocytes to turn into neurons. In mouse fashions of Parkinson’s illness, Fu’s crew discovered that animals that acquired the therapy confirmed enchancment in sure behaviours.
Cleveland, who was concerned in Fu’s trial, has been working with an ASO equipped by Ionis to check the concept in different elements of the brain. “This is really where I’m going to invest the rest of what I’ve got left as a career,” he says. “I’m confident that we have only begun to think about the possibilities.”
These astrocyte-converting ASOs are nonetheless at an early stage. Fu cautions that earlier than this system is taken to the clinic, it must be examined in non-human primates, as a result of their brains match our personal extra carefully than do these of mice.
For now, researchers are eagerly awaiting the outcomes of the tofersen part III trials in ALS, and for extra details about precisely why the tominersen trial for Huntington’s was halted.
Susan, a retired nurse in her mid-60s, has been concerned within the tominersen trial since part I. She is upset within the information, she says, however is grateful for the care she’s acquired as a participant. “I’ve been so privileged to be part of this trial right since day one. Now it’s just about patience and reviewing. There’s no alternative, is there?”