{kind=link}
Animals
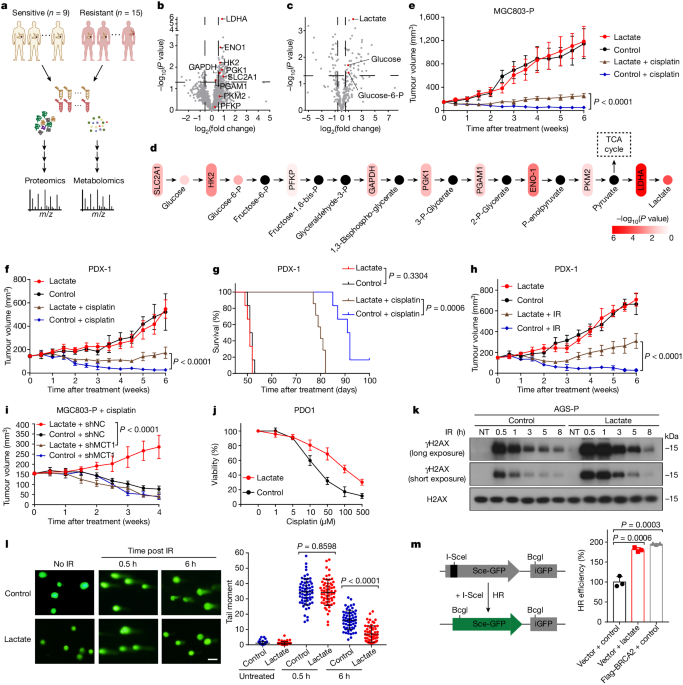
All animal studies were performed in accordance with the Animal Care and Use Committee of Sun Yat-sen University. For all mouse experiments (including PDXs), the maximum permitted tumour volume below 1,600 mm3 was not exceeded. Mice were kept under specific pathogen-free or germ-free conditions, with an ambient temperature of 20 ± 2 °C, humidity of 55 ± 10% and a dark:light cycle of 12 h. Six-week-old male NSG mice were allowed to acclimatize to housing conditions in animal facility for 1 week before being used in the experiments. Both male and female mice were used for experiments, but within each experiment, they were sex-matched. Cells were resuspended in 1:1 PBS:Matrigel and subcutaneously transplanted into the bilateral dorsal flanks of NSG mice. The subcutaneous PDX model was established by transplanting minced fresh tumour tissue into NSG mice.
In animal experiments involving lactate treatment, the mice were assigned randomly to the following groups: (1) control (saline); (2) lactate (100 μl of 1 mM, 3 times a week); (3) cisplatin (2 mg kg−1, once a week) or IR (2 Gy per fraction, once daily for 4 consecutive days per week); (4) a combination of both agents at the aforementioned doses (n = 6 mice per group). Sample size in each group was determined by our preliminary experiments. For cisplatin and lactate administration, mice in the treated groups received intraperitoneal injections.
In animal experiments involving stiripentol treatment, mice were treated as follows: (1) control (saline); (2) IR (2 Gy per fraction, once daily for consecutive 4 days per week) or cisplatin (2 mg kg−1, once a week); (3) stiripentol (150 mg kg−1, once daily for consecutive 5 days per week); (4) the combination of both agents at the aforementioned doses. For cisplatin and stiripentol administration, mice in the treated groups received intraperitoneal injections. Tumour volume and body weight were measured every three days weekly. Tumour volume was calculated using the following formula: volume (mm3) = [width (mm)]2 × length (mm)/2.
Organoid cultures
Gastric cancer organoids were established as described27. In brief, gastric cancer tissue used for organoid culture was obtained following surgery from patients with gastric cancer. Tumour tissues were isolated and transported to the laboratory on ice within 1 h of removal from the patients in ice-cold DMEM/F-12 with 50 U ml−1 penicillin-streptomycin. Tissues were washed three times with cold DMEM/F-12 with antibiotics and cut into small pieces with sterile blades. The minced tissue was incubated in DMEM containing 1 mg ml−1 collagenase V (Sigma) for 1 h at 37 °C. The tissues were washed in ice-cold PBS, followed by centrifugation (300g, 5 min, and 4 °C). TrypLE (Thermo Fisher Scientific) was used to digest the sample for 5 min at 37 °C, followed by stopping with ice-cold PBS and centrifugation. The sample was resuspended in 50 μl culture medium and then filtered through a 40-μm nylon mesh. One-hundred microlitres Matrigel (Corning) was added to the suspension, which was allowed to solidify on pre-warmed 24-well culture plates (Corning) for 15 min at 37 °C. One millilitre culture medium was added to the well after gelation. The medium was changed every 3–4 days, and the organoids were passaged with TrypLE every 2 weeks. The medium for culturing gastric cancer organoids was as described previously28.
Cell lines
Cell lines were cultured in a humidified incubator at 5% CO2 and 37 °C. All cell lines were validated by STR DNA profiling and tested negative for mycoplasma by PCR. Culture media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
293 T, AGS, A549, HCT116 and HeLa cell lines were obtained from American Type Culture Collection (ATCC). MGC803 cells were obtained from Cell Bank, Shanghai Institute of Biochemistry and Cell Biology (SIBCB). U2OS-265 cells were provided by R. Greenberg. 293 T and HeLa cells were cultured in DMEM medium (Gibco). AGS and A549 cells were cultured in F12K medium (Gibco). HCT116, U2OS-265 and MGC803 cells were cultured in RPMI 1640 medium (Gibco).
Patients and tumour samples
Tumour samples from patients with gastric cancer, pathologically and clinically diagnosed at the Seventh Affiliated Hospital of Sun Yat-sen University, were collected. Informed consent was obtained from all patients, and approvals were obtained from the ethics board of the Seventh Affiliated Hospital of Sun Yat-sen University for the use of these specimens in research. The Institutional Review Board or IRB (Number KY-2022-011-01 and KY-2022-039-02) at the Seventh Affiliated Hospital of Sun Yat-sen University. Clinical information on these patients, including age, chemotherapy regimens, and survival situation, was obtained from medical and follow-up records (Supplementary Tables 5 and 6). Pathological tumour regression grade (TRG) was used to evaluate the efficacy of NAC. TGR was classified into four tiers according to Ryan’s score29. Scores of 0 to 2 were categorized as responders or sensitive, whereas score 3 was categorized as non-responders or resistant.
Antibodies
The following antibodies were generated by Cell Signaling: anti-NBS1 (14956); anti-caspase-3 (14220); anti-H2AX (7631); anti-H2AX (Ser139) (9718); anti-histone H3 (4620); anti-p300 (86377); anti-HDAC3 (3949); anti-histone H3 (4499). The following antibodies were generated by Novus: anti-NBS1 (NB100-143SS). The following antibodies were generated by ABclonal: anti-Flag (AE005); anti-β-actin (AC004). The following antibodies were generated by Proteintech: anti-β-tubulin (10068-1-AP); anti-MCT1 (20139-1-AP); anti-LDHA (19987-1-AP); anti-TIP60 (10827-1-AP); anti-GFP (50430-2-AP); anti-c-MYC (10828-1-AP). The following antibodies were generated by BD: anti-H2AX (pS139) (560446); anti-RAD50 (611010). The following antibodies were generated by Abcam: anti-RAD51 (ab88572); anti-TIP60 (ab300522); anti-histone H4 (ab31830). The following antibodies were generated by PTM BIO: anti-pan-Kla (PTM-1401); anti-pan-Kac (PTM-101); anti-histone H4K8ac (PTM-120); anti-NBS1-K388la (N/A). The following antibodies were generated by Santa Cruz: anti-BRCA1 (sc-6954).
For western blots, antibodies were diluted 1:1,000. For immunofluorescence, antibodies were diluted 1:200. For IHC, antibodies were diluted 1:100.
Prime editing-mediated genome editing
The prime editing system was used to construct genomic NBS1(K388R) mutations in AGS parental cells. Prime editing was performed as described previously30. In brief, the pegRNA-NBS1 spacer sequence and 3′ extension sequence were designed using the prime-editing guide RNAs design tool (http://pegfinder.sidichenlab.org/). Prime editing-NBS1 spacer sequence: GAAATCAAAGTCTCCAAAA. Prime editing-NBS1 3′ extension sequence: TTTTTGTTCCATTCTGGAGACTTTGAT. The digested pU6-pegRNA-GG-vector plasmid was assembled with the spacer sequence, 3′ extension sequence, and scaffold sequence by Golden Gate assembly. The ligation product was transformed into Escherichia coli. The resulting clonal transformants were isolated and sequenced. PCMV-PE2 and assembled pU6-pegRNA-GG-Vector plasmid were transfected into AGS parental cells. After 24 h post-transfection, cells were diluted and seeded into 96-well plates with only one cell per well. After cultivation, Genomic DNA was then extracted from the monoclonal cells. The PCR products spanning the mutation sites were sequenced.
PDX model
The collection of gastric cancer tumour surgical specimens was approved by the Seventh Affiliated Hospital of Sun Yat-sen University. The informed consent of patient was obtained according to institutional regulatory standards before surgery. Tumour tissues were collected, and transported to the laboratory within 1 h in ice-cold DMEM with 50 U ml−1 penicillin-streptomycin. Tumour tissues were washed three times with cold DMEM with 50 U ml−1 penicillin-streptomycin and cut into small pieces with sterile blades. A small incision was made on the bilateral dorsal flanks of anaesthetized NSG mice and minced fresh tumour surgical specimens were subcutaneously transplanted. The incision was closed up with sutures and tumour formation was monitored for the next 3 months.
Neutral comet assays
Neutral comet assays were performed using the Comet Assay Kit (Trevigen) according to the manufacturer’s protocol. In brief, the lysis solution was prepared and chilled at 4 °C for at least 20 min before use. Agarose was melted in a water bath of boiling water for 5 min and then cooled in a 37 °C water bath for at least 20 min. Cells (1 × 105 ml−1) were combined with molten agarose at a ratio of 1:10 (v/v), and 50 µl was placed onto the comet slide. The slides were placed in a 4 °C refrigerator for 10 min, and then were immersed in a 4 °C lysis solution for 1 h, followed by neutral electrophoresis buffer for 30 min. The slides were subjected to electrophoresis at 21 V for 45 min and immersed in DNA precipitation solution and 70% ethanol for 30 min at room temperature. The samples were dried at 37 °C for 10 min and stained with SYBR green for 10 min before the images were captured under an epifluorescence microscope (Olympus). The tail moment was analysed using the Comet Assay Software Project (CASP).
HR and NHEJ reporter assays
HeLa cells were stably integrated with DR-GFP (Addgene, #26475) and EJ5-GFP (Addgene, #44026) reporters respectively. I-SceI-T2A-dsRed was a gift from L. Li. In brief, 1 × 106 HeLa DR-GFP or EJ5-GFP reporter cells were transfected with 3 μg of I-SceI using Lipofectamine 3000 Transfection Kit (Invitrogen). After 48 h, cells were collected and subjected to flow cytometry analysis (CytoFLEX). The efficiency of repair was determined by the ratio of cells expressing both GFP and dsRed signals to all dsRed-positive cells. Three independent experiments were performed.
Laser microirradiation, imaging and immunofluorescence
Cells were transfected with the indicated GFP-tagged expression plasmids and seeded onto 35-mm glass-bottom dishes (NEST). After 24 h transfection, cells were placed into a cell culture chamber (37 °C, 5% CO2) on an inverted microscope (Olympus). Laser microirradiation was carried out by scanning the regions of interest using fixed wavelength of ultraviolet laser (405 nm). Time-lapse images were captured and the fluorescence intensity of the micro-irradiated regions within the nucleus relative to the non-irradiated regions was calculated using Olympus software.
For immunofluorescence assay, cells were seeded into glass-bottom dishes (NEST), and then treated with cytoskeleton buffer (10 mM PIPES, pH 7.0, 100 mM NaCl, 300 mM sucrose, and 3 mM MgCl2, 0.7% Triton X-100, 0.3 mg ml−1 RNase A) for 5 min. Next, cells were fixed with 4% (w/v) paraformaldehyde (Sigma) for 15 min at room temperature, washed with 1× PBS 3 times. Cells were permeabilized with 0.2% Triton X-100 for 5 min and blocked in immunostaining blocking solution (Beyotime) for 30 min. Subsequently, the cells were washed three times with PBS and then incubated with the indicated primary antibody at 4 °C overnight. Finally, images were captured with a fluorescence microscope (Olympus or Leica).
Establishment of cisplatin-resistant cell lines
For cancer cell lines (AGS, MGC803, HCT116, HGC27 and A549), cells reaching approximately 70% density in 100 mm dishes were treated with one-tenth of the half-maximal inhibitory concentration (IC50) of cisplatin. Fresh drug-containing complete culture medium was then changed every two days. The cells were treated with sequentially increasing concentrations of cisplatin for nearly six months. The IC50 values of the cisplatin-resistant cells were at least 5-fold higher than those of the corresponding parent cells.
Measurement of lactate
To measure lactate levels in tumour tissue, the tissue was homogenized with lysis buffer on ice, and the supernatant was obtained by centrifugation at 12,000g for 10 min at 4 °C. The supernatants were collected, and lactate levels were measured using an l-Lactate Assay Kit (Abcam, ab65330) following the manufacturer’s instructions.
Crosslinking mass spectrometry
AGS-P cells and AGS-NBS1(K388R) cells were grown to 80% confluence, lysed with NETN buffer, and clarified via centrifugation. To enrich NBS1-interacting proteins, 5 μg anti-NBS1 antibody was incubated with 40 μl protein A/G beads at room temperature for 2 h. After washing twice, the beads were added to 1 mg of cell lysate and incubated at room temperature for 2 h. Proteins bound to beads were resuspended with HEPES buffer, added with DSSO at 2.5 mM final concentration, and crosslinked at room temperature for 1 h with shaking. Subsequently, 1 M Tris-HCl (pH 8.0) was added to a final concentration of 62 mM to quench the crosslinked reaction. The crosslinked NBS1-interacting proteins were reduced with 50 mM dithiothreitol at 37 °C for 1.5 h, alkylated with 50 mM iodoacetamide for 15 min at room temperature in darkness, and digested with 1 μg trypsin at 37 °C overnight. After desalination, the crosslinked peptides were analysed by LC–MS/MS and identified through database searching, as previously described.
4D label-free quantitative lactylproteomics analysis
Cells were collected, and the 4D label-free quantitative lactylproteomics analysis was performed by Jingjie PTM BioLabs. For protein extraction, cell sample was grinded by liquid nitrogen, and then the powder was sonicated three times in lysis buffer (50 µM PR-619, 1% Triton X-100, 50 mM NAM, 10 mM dithiothreitol, 1% protease inhibitor cocktail, 3 µM trichostatin A (TSA) and 2 mM EDTA) on ice using a high-intensity ultrasonic processor (Scientz). An equivalent volume of Tris-saturated phenol was added to the sample, which was then vortexed for 5 min. The upper phenol phase was transferred to a new tube tube after centrifugation (4 °C, 10 min, 5,000g). Proteins were precipitated by adding at least four volumes of ammonium sulfate-saturated methanol. The mixture was further incubated for at least 6 h at −20 °C. The supernatant was discarded after centrifugation (4 °C, 10 min). The remaining precipitate was washed once with ice-cold methanol and then three times with ice-cold acetone. Proteins were re-dissolved with 8 M urea and the protein concentration was measured with a BCA kit.
For digestion, the protein solution was reduced with 5 mM dithiothreitol for 30 min at 56 °C, and was alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. Subsequently, 100 mM triethylammonium bicarbonate (TEAB) was added to urea in the protein sample that was then digested overnight by trypsin at 1:50 trypsin-to-protein mass ratio.
To enrich lactyl-modified peptides, tryptic peptides were dissolved in NETN buffer (1 mM EDTA, 100 mM NaCl, 50 mM Tris-HCl, 0.5% NP-40, pH 8.0) and incubated with pre-washed antibody beads at 4 °C overnight with gentle shaking. The breads were washed four times with NETN buffer and twice with water. The peptides were eluted from the beads with 0.1% TFA and then vacuum-dried.
For LC–MS/MS analysis, tryptic peptides were dissolved in solvent A (2% acetonitrile in water and 0.1% formic acid) and then loaded onto a home-made reversed-phase analytical column. Peptides were separated with a gradient from 6% to 24% solvent B (0.1% formic acid in acetonitrile) in 70 min, 24% to 35% over 14 min, 35% to 80% over 3 min, and held at 80% for the last 3 min, all at a constant flow rate of 450 nl min−1 using a nanoElute UHPLC system (Bruker Daltonics). The peptides were subjected to capillary source and analysed using the timsTOF Pro (Bruker Daltonics) mass spectrometry. The timsTOF Pro was operated in parallel accumulation serial fragmentation (PASEF) mode. Precursors and fragments were analysed on TOF detector (a MS/MS scan range from 100 to 1,700 m/z). The dynamic exclusion was set to 30 s. Precursors with charge states 0 to 5 were selected for fragmentation, and 10 PASEF-MS/MS scans were acquired per cycle. The electrospray voltage applied was 1.75 kV.
For database search, the MS/MS data were processed with Maxquant search engine (v.1.6.6.0). Tandem mass spectra were searched against the Homo_sapiens_9606_SP_20200509. Fasta concatenated with reverse decoy database. Trypsin/P was specified as cleavage enzyme allowing up to 2 missing cleavages. The mass tolerance for precursor ions was set as 20 ppm in main search and 20 ppm in first search and the mass tolerance for fragment ions was set as 0.04 Da. Carbamidomethyl on Cys was specified as fixed modification, and lactylation on Lys and oxidation on Met were specified as variable modifications. False discovery rate was adjusted to <1%.
Label-free proteomics analysis
For tumour tissue, the tissue was washed wash away the remaining blood and other body fluids on the tissue surface by using PBS. The tissue was cut with scissors, and sonicated 3 times in lysis buffer (50 µM PR-619, 1% Triton X-100, 50 mM NAM, 10 mM dithiothreitol, 1% protease inhibitor cocktail, 3 µM TSA and 2 mM EDTA) on ice using a high-intensity ultrasonic processor (Scientz). An equivalent volume of Tris-saturated phenol was added to the sample, which was then vortexed for 5 min. The upper phenol phase was transferred to a new tube and centrifuged (4 °C, 10 min, 16,000g). Proteins were precipitated by adding at least four volumes of ammonium sulfate-saturated methanol. The mixture was further incubated for at least 6 h at −20 °C. The supernatant was discarded after centrifugation (4 °C, 10 min). The remaining precipitate was washed once with ice-cold methanol and then three times with ice-cold acetone. Proteins were re-dissolved with 8 M urea and the protein concentration was measured with a BCA kit. Proteins were reduced with 5 mM dithiothreitol at 37 °C for 1 h. Proteins were alkylated with 10 mM iodoacetamide at 25 °C for 45 min in the dark. Samples were digested with trypsin (Promega) at 1:50 enzyme-to-protein ratio. After 18 h of digestion, peptides were eluted with 0.1% TFA and vacuum-dried. Peptides were analysed by LC–MS/MS (Thermo Fisher Easy1200-Faims Fusion Orbitrap).
For cell samples, cells were lysed with lysis buffer (50 mM Tris-HCl [pH 8.0], 1% Triton X-100, 0.5% Nonidet P-40, 10 mM dithiothreitol, 1% protease inhibitor cocktail, 150 mM NaCl and 5 mM EDTA) on ice for 30 min, followed by centrifugation (12,000g, 20 min, and 4 °C). The protein solution was precipitated with acetone, and was reduced with 50 mM dithiothreitol for 1.5 h at 30 °C. The protein solution was alkylated with 50 mM iodoacetamide for 15 min at room temperature in darkness. Subsequently, 100 mM TEAB was added to urea in the protein sample that was then digested overnight by trypsin at 1:50 trypsin-to-protein mass ratio. Finally, the peptides were analysed by LC–MS/MS (Thermo Fisher Easy1200-Faims Fusion Orbitrap).
Metabolomics analysis
Tumour tissue were pulverized after being frozen in liquid nitrogen with the addition of 250 µl of mixed solvent (chloroform:methanol:water, 1:2:1). The lysate was sonicated and centrifuged for 10 min at 12,000 rpm. Aqueous supernatant was transferred to a gas chromatography vial containing internal standards. The deposit was rehomogenized with a T10 basic homogenizer at 4 °C for 30 s after adding 250 µl of methanol. An aliquot of supernatant was added to the mixture in the vial and vacuum-dried after a second centrifugation. Samples were run on an LC–MS/MS (Thermo Fisher Ult3000-Exploris 480 Orbitrap).
Quantification and statistical analysis
All statistical analyses were performed with GraphPad Prism 7.0 (GraphPad). Values were obtained from at least three independent experiments, using three technical replicates per condition, unless otherwise indicated in the figure legend. No animals or tumour samples were excluded from data analyses. Student’s t-test, two-sided, unpaired, two-tailed, two-way or one-way analysis of variance (ANOVA) were used to analyse data as indicated. The Kaplan–Meier method was used to calculate the cumulative overall survival data, and the log-rank test was used for analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.